Back To Listings

- First authors: Young-Han Shin
- Corresponding authors: Andrew M. Rappe
- Whole authors: Young-Han Shin, Valentino R. Cooper, Ilya Grinberg, Andrew M. Rappe
- Authors from M3L: Young-Han Shin
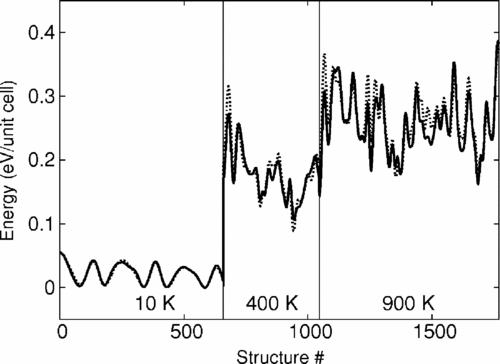
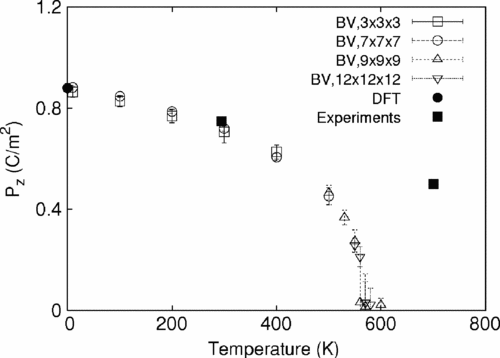
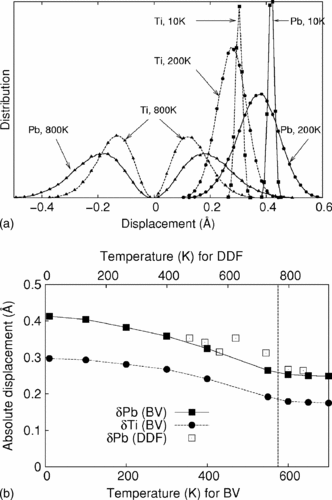
A simple ten-parameter interatomic potential model is described that is capable of accurately reproducing the static and dynamical properties of complex oxides. The accuracy of this model stems from the crystal-chemical bond-valence theory of ionic and covalent bonding. The development of a specific variant of this model for ferroelectric PbTiO3 (PT) is discussed in detail, and comparison of the model is made with density functional theory computations and with experimental data. Bond-valence molecular dynamics (BVMD) simulations for PT show a ferroelectric transition at 575 K. The BVMD model correctly reproduces the mixed order-disorder and displacive phase transition character, the magnitudes of cation displacements in the ferroelectric and paraelectric phases, and the energy of 180° domain walls. The success of this simple and physically motivated model makes the simulation of extended defects tractable in PT and other complex oxides.
Authors from M3L