Back To Listings

- First authors: Ilya Grinberg
- Corresponding authors: Andrew M. Rappe
- Whole authors: Ilya Grinberg, Young-Han Shin, Andrew M. Rappe
- Authors from M3L: Young-Han Shin
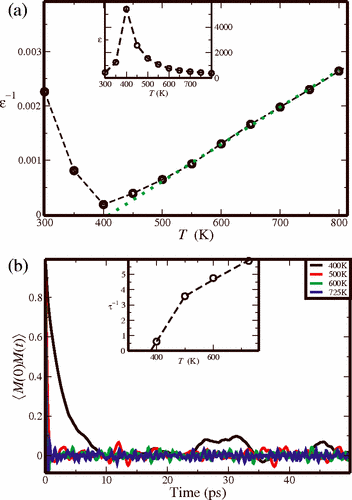
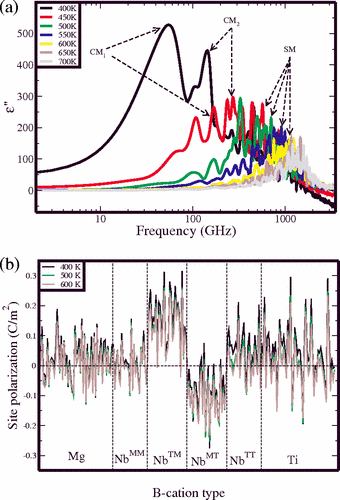
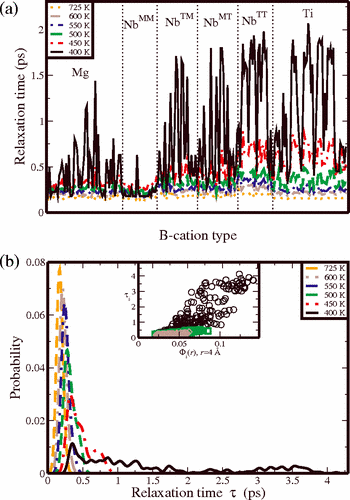
We use atomistic molecular dynamics simulations to study relaxor behavior in the
0.75PbMg1/3Nb2/3O3-0.25PbTiO3 material. Even for a fairly small simulation size of 1000 atoms, the system exhibits frequency dispersion and deviation from the Curie-Weiss law typical of relaxor materials. Analysis of the time autocorrelation functions for individual atoms allows us to identify the Nb atoms with a high concentration of neighboring Ti atoms as the nucleation sites for the relaxor behavior. This is due to the higher coupling between the cation displacements induced by the presence of overbonded oxygen atoms.
Authors from M3L