Back To Listings

- First authors: Won Joon Heo
- Corresponding authors: Eok Kyun Lee
- Whole authors: Won Joon Heo, Won-June Kim, Young-Han Shin, Eok Kyun Lee
- Authors from M3L: Young-Han Shin
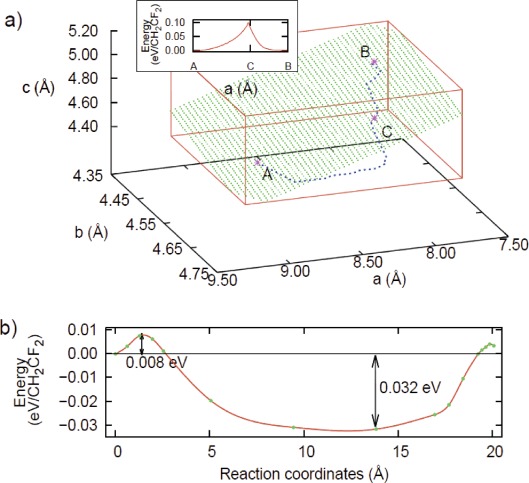
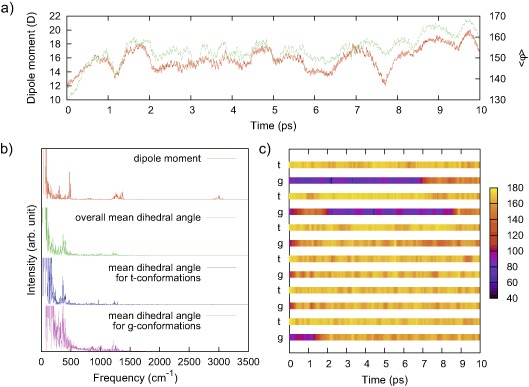
We perform density functional calculations to investigate structural and dynamical properties of crystalline polyvinylidene difluoride (PVDF) associated with the transition from α to β phase. We examine the change of the conformational energy and the corresponding structure of each phase depending on the lattice parameters of the orthorhombic crystalline structure. From this information, we construct the path that connects the point where the α phase is most stable to the point where the β phase is most stable, and identify the sub- region in the lattice parameter space where α and β phases have the same energy. In this sub-region, we locate the point which gives the lowest conformation energy for both α and β phases, and examine the behaviour of the lowest energy profile and corresponding change of intermediate structures as the conformation of the PVDF chain transforms from α phase to β phase. Finally we perform ab-initio molecular dynamics simulations and analyse the characteristic dynamics associated with transition from α to β phase.
Authors from M3L